 热点资讯
热点资讯甲胎蛋白持续升高, 肝功能持续异常, 不是脂肪肝和自免肝
患者,男性,两年前找我看肝病,当时46岁左右。
转氨酶不高,在80左右,但是谷氨酰转肽酶明显升高,300左右,持续服用熊去氧胆酸几乎纹丝不动。总胆红素偏高。
乙肝两对半显示单纯核心抗体阳性,B超提示脂肪肝,肝脏纤维化扫描提示肝脏硬度值14.3 Kpa、脂肪衰减301 (重度脂肪肝)。
建议患者继续服用熊去氧胆酸和积极减肥,其实患者并不胖,我想当然认为是所谓的“瘦人脂肪肝。”
患者体重减轻后肝功能仍然异常,并没有丝毫改变。这让我推翻了自己“瘦人脂肪肝”的诊断。
转而考虑是否为自身免疫性肝病,完善了相关自身抗体的检查,提示均为阴性。
后该患者去上海就诊,门诊医生也没有做出明确诊断。我建议患者尽快做一次肝活检,以获得病理学的诊断,该患者始终犹豫。
曾经尝试过激素治疗,效果不佳。后该患者找其他消化科医生看病,几个月后发现腹水。
再次去上海住院治疗,通过抽血做基因检查和24小时尿铜等检查,明确诊断是肝豆状核变性。
揭开迷雾:什么是肝豆状核变性?
上面这位患者曲折的就诊经历,最终指向了一个并不十分常见、却极易被误诊的疾病——肝豆状核变性。下面我们就来系统认识一下这种“伪装大师”般的疾病。
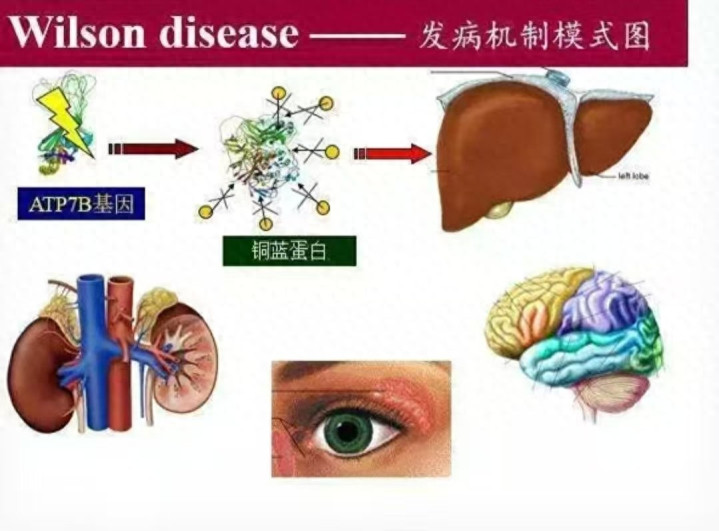
一、病因:一个铜代谢的“程序错误”
肝豆状核变性(又称Wilson病)是一种常染色体隐性遗传病。简单来说,患者从父母双方各继承了一个有缺陷的基因(ATP7B基因)。这个基因原本负责编码一种“铜转运蛋白”,帮助肝脏把多余的铜排入胆汁,从而排出体外。
基因突变后,这个“排铜系统”失灵了。铜无法正常排泄,便在体内逐渐堆积,首先攻击肝脏,随后侵犯大脑、角膜、肾脏等器官。
二、发病机制:铜过量,步步惊心
正常人体内铜含量约为50-100毫克,每天通过饮食摄入的铜需要靠胆汁排出以维持平衡。肝豆状核变性患者由于排铜障碍,铜像“垃圾”一样在肝细胞内越积越多:
· 早期:铜沉积引起肝细胞脂肪变性、炎症、坏死,表现为转氨酶升高、脂肪肝(即使人不胖)、肝纤维化。
· 中期:肝损伤持续,可发展为肝硬化,甚至出现腹水、黄疸、食管静脉曲张。
· 后期:铜溢出肝脏进入血液,沉积在大脑基底节区,引起神经精神症状(如肢体震颤、说话含糊、性格改变);沉积在角膜形成特征性的K-F环(铜色色素环);沉积在肾脏导致肾小管损伤。
三、发病特点:善于伪装的“变色龙”
肝豆状核变性可以在任何年龄发病,但多见于5-35岁。它的表现极其多样,因此极易误诊:
常见表现类型 具体症状
肝脏型 类似脂肪肝、自身免疫性肝炎、不明原因的转氨酶/谷氨酰转肽酶持续升高、肝硬化、腹水,甚至急性肝衰竭。
神经型 手脚不自主抖动、写字变小、口齿不清、流口水、走路不稳、面具脸。
精神型 情绪波动、抑郁、焦虑、学习成绩下降、行为怪异,常被误诊为心理疾病。
其他 溶血性贫血(铜释放入血破坏红细胞)、关节疼痛、肾结石等。
与本文病例呼应:该患者表现为持续转氨酶升高、谷氨酰转肽酶显著升高、熊去氧胆酸无效、激素治疗无效、最终出现腹水——这完全是铜沉积导致肝硬化的典型过程,却先后被误认为“瘦人脂肪肝”和“自身免疫性肝病”。临床上,对于不明原因的慢性肝病,尤其是伴有神经症状或家族史的患者,一定要警惕肝豆状核变性。
四、诊断:如何抓住这个“隐形杀手”?
确诊肝豆状核变性并不困难,关键是要想到它。常用检查包括:
1. 血清铜蓝蛋白:绝大多数患者显著降低(
2. 24小时尿铜排泄量:>100 μg有诊断价值(本文患者正是通过此检查确诊)。
3. 角膜K-F环:裂隙灯下检查,是神经型患者的典型标志。
4. 肝脏铜含量:肝活检组织检测,>250 μg/g干重是金标准之一。
5. 基因检测:ATP7B基因突变分析,可明确诊断并用于家族筛查。
五、治疗与转归:早期治疗,预后良好
肝豆状核变性是少数可治疗的遗传代谢病之一。一旦确诊,必须终身治疗,但规范治疗后绝大多数患者可以正常生活。
治疗原则:排铜+限铜
· 驱铜药物:
· 青霉胺:经典排铜剂,但副作用较多(过敏、肾损伤等)。
· 二巯基丙醇/曲恩汀:效果较好,副作用相对少。
· 锌剂:通过抑制肠道对铜的吸收来“堵住源头”,适合维持治疗或无症状患者。
· 低铜饮食:
· 避免:动物肝脏、贝壳类海鲜、坚果、巧克力、蘑菇、豆类。
· 适宜:精白米面、瘦肉、鸡蛋、牛奶、大部分蔬菜水果。
· 注意:不要使用铜制器皿烹饪或储存酸性食物。
· 对症支持:肝硬化并发症(腹水、出血)需相应处理;神经症状可辅以康复治疗。
预后与转归
· 早期(肝硬化前)开始治疗:肝功能可完全恢复正常,肝脏纤维化甚至可逆转,患者能正常工作和生活。
· 中期(代偿期肝硬化):坚持排铜治疗可稳定病情,延缓进展,多数患者预后良好。
· 晚期(失代偿期肝硬化或急性肝衰竭):药物治疗效果差,可能需要肝移植。肝移植不仅能纠正肝硬化,还能提供正常的ATP7B基因,从根本上治愈该病。
· 神经症状:治疗开始后数月至数年,多数患者的震颤、言语障碍可明显改善,但部分严重损伤可能残留。
重要提醒:本病治疗不可中断!擅自停药会导致铜快速再沉积,病情急剧恶化。同时,患者的兄弟姐妹和子女应进行基因筛查,及早发现无症状的患者,早治疗可完全避免发病。
总结与启示
回到本文开头的病例:一位中年男性,不胖却有重度脂肪肝,转氨酶轻度升高但谷氨酰转肽酶显著升高,熊去氧胆酸和激素均无效,最终发展为腹水——这些“不合常理”的线索,恰恰指向了肝豆状核变性。如果当初能更早想到这个病,完成24小时尿铜或基因检查,患者或许可以避免走向肝硬化和腹水。
给医生和患者的几点忠告:
· 对于任何年龄的不明原因肝病,尤其伴有神经症状、精神异常或溶血时,请把肝豆状核变性列入鉴别诊断。
· 单纯“脂肪肝”但患者不胖,或减肥后肝功能纹丝不动——请警惕遗传代谢病。
· 自身免疫性肝病相关抗体阴性,激素治疗无效——不要一味坚持原诊断,及时拓宽思路。
· 24小时尿铜、铜蓝蛋白、裂隙灯查K-F环,是性价比极高的筛查手段。
肝豆状核变性虽为遗传病,但绝非不治之症。早诊断、早排铜、终身管理,患者完全可以拥有与健康人无异的人生。希望这篇科普能帮助更多人认识这个“伪装大师”,少一些误诊,多一些康复。
